
Imagine spending eight hours in bed and waking up more exhausted than when you went to sleep. You’re getting plenty of time under the covers, but somehow your body never gets the rest it desperately needs. For millions of Americans, this isn’t just an occasional bad night—it’s a nightly battle with a condition called sleep apnea, where breathing repeatedly stops and starts throughout the night.
Sleep apnea causes repeated interruptions in breathing during sleep, and it’s far more common than most people realize. Over 936 million adults worldwide aged 30-69 have some form of sleep apnea, though many don’t know they have it. The challenge is that the most dramatic symptoms happen while you’re unconscious, making it a disorder that often requires someone else to notice before you realize there’s a problem.
What Actually Happens During Sleep Apnea
The most common form is obstructive sleep apnea, which accounts for the vast majority of cases. During obstructive sleep apnea, the muscles in the back of the throat relax too much, causing the upper airway to collapse and interrupt breathing. Think of it like trying to breathe through a straw that keeps getting pinched shut—your body is trying to get air, but the pathway keeps getting blocked.
Each pause in breathing, called an apneic event, lasts at least 10 seconds and can occur dozens or even hundreds of times per night. When oxygen levels drop, your brain essentially sounds an alarm, triggering a brief arousal to restart breathing. These micro-awakenings fragment your sleep into tiny pieces, preventing you from getting the deep, restorative rest your body needs. The termination of each apneic event is associated with a transient arousal from sleep, but these don’t register in your conscious memory. You don’t wake up in the morning thinking “I woke up 247 times last night.” Instead, you just feel terrible despite thinking you slept through the night.
There’s also central sleep apnea, which works differently. With central sleep apnea, the brain fails to send proper signals to the breathing muscles during sleep. Instead of a physical blockage, it’s a communication breakdown between your brain and the muscles that control breathing. Some people even have complex sleep apnea, which combines both types.
How Sleep Apnea Gets Diagnosed
Doctors measure the severity of sleep apnea using the apnea-hypopnea index, which counts the average number of breathing pauses per hour. An AHI between 5 and 14 events per hour is considered mild, 15 to 29 is moderate, and 30 or more is severe. Someone with severe sleep apnea might stop breathing more times in a single night than they’d care to count.
The gold standard for diagnosis is polysomnography—an overnight sleep study conducted in a specialized lab. During polysomnography, doctors monitor brain waves through an electroencephalogram, track oxygen levels with pulse oximetry, measure airflow through temperature and pressure sensors, and use respiratory belts to monitor chest and abdomen movement. It’s like turning your body into a living data collection system for one night, with sensors measuring everything from your heart rate to how your legs move during different sleep stages.
For many people, though, home sleep testing is a more convenient option. Home sleep apnea tests use a limited subset of measurements—typically heart rate, oxygen levels, respiratory effort, body position, and nasal airflow. While these portable devices can’t capture everything a full lab study can, they’re often sufficient for diagnosis and much easier to arrange.
Treatment Options: From Lifestyle Changes to High-Tech Solutions
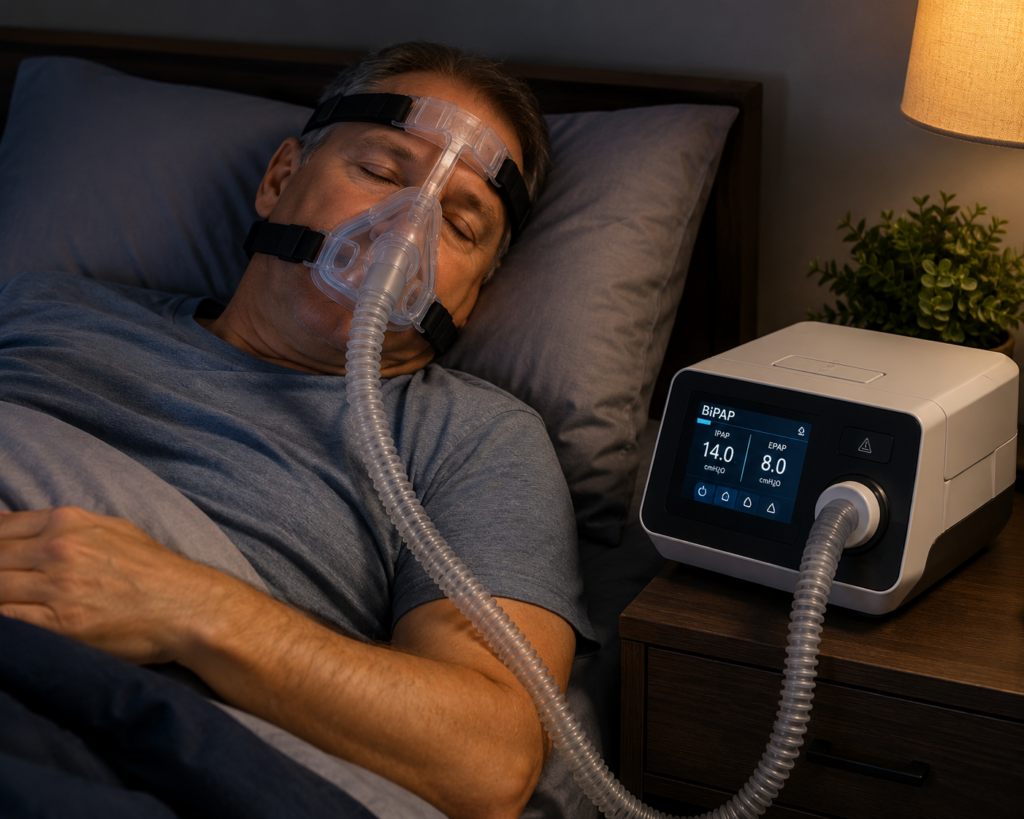
The most effective treatment for moderate to severe sleep apnea is continuous positive airway pressure therapy, better known as CPAP. CPAP works by delivering air through a mask to keep airways open during sleep. Think of it as a gentle wind tunnel that prevents your throat from collapsing. The device maintains constant air pressure, essentially splinting your airway open so you can breathe normally throughout the night.
But here’s the challenge: nearly half of patients don’t consistently use CPAP after the first month. The mask can be uncomfortable, some people find the pressure sensation strange, and it takes getting used to sleeping with equipment on your face. For people who can’t tolerate CPAP or have milder cases, there are alternatives like oral appliances that reposition the jaw and tongue to keep the airway open.
Surgery is another option, though it’s typically reserved for specific situations. Surgical options include procedures to remove enlarged tonsils or adenoids, or to address abnormalities causing airway obstruction. There’s even a newer treatment called hypoglossal nerve stimulation, which uses an implanted device to stimulate the nerve controlling tongue movement, keeping it from blocking the airway.
For some people, lifestyle modifications can make a real difference, especially with mild cases. Weight loss often helps since obesity is a major risk factor for sleep apnea. Sleeping on your side instead of your back can reduce airway collapse, and avoiding alcohol before bed helps because alcohol relaxes throat muscles even more.
Sleep Apnea Versus Insomnia: Two Very Different Beasts
Here’s where things get interesting—and sometimes confusing. Sleep apnea and insomnia are fundamentally different disorders, yet they can look similar and even occur together. Sleep apnea is primarily a problem of abnormal breathing during sleep, while insomnia is an inability to fall asleep or stay asleep.
With insomnia, your brain is often the problem—it won’t shut off when you want it to. You lie awake with racing thoughts, or you fall asleep only to pop awake an hour later, mind already churning. People with insomnia are usually very aware that they’re having sleeping difficulties. They know exactly how long they’ve been staring at the ceiling.
Sleep apnea operates differently. Many people with sleep apnea don’t realize they have breathing problems during sleep, as the awakenings are so brief they’re not consciously aware of them. You might spend what feels like a full night asleep yet wake up exhausted. Your problem isn’t falling or staying asleep in the traditional sense, it’s that the sleep you’re getting is constantly interrupted and of terrible quality.
The plot twist is that these conditions can feed into each other. Sleep apnea causes numerous micro-awakenings that disrupt sleep and can lead to insomnia-like symptoms. When you’re waking up dozens or hundreds of times per night, even if you don’t fully remember it, your sleep architecture gets shredded. This fragmentation can trigger the hyperarousal and sleep anxiety characteristic of insomnia.
Between 30-50% of people with sleep apnea also experience chronic insomnia symptoms, a combination researchers call COMISA (comorbid insomnia and sleep apnea). This overlap creates treatment challenges because patients with both conditions may have difficulty tolerating CPAP due to frequent awakenings and an inability to fall back asleep with the mask in place.
How Would You Know If You Have Sleep Apnea?
This is the tricky part—you’re unconscious when it happens. Loud snoring, snorting, or gasping during sleep is a major warning sign, though not everyone who snores has sleep apnea and not everyone with sleep apnea snores. If you live alone, you might never hear these sounds yourself.
Often, it’s a bed partner who first notices something’s wrong. They might observe you stop breathing, then suddenly gasp or snort as you restart. People with sleep apnea often toss and turn and show signs of restless nighttime sleep, sometimes waking up with sheets twisted and pillows scattered.
The daytime symptoms are easier to recognize. Waking with a headache, not feeling refreshed despite adequate time in bed, and feeling tired throughout the day, even to the point of dozing off, can all signal sleep apnea. If you find yourself nodding off while reading, watching TV, or worst of all, driving, that’s a red flag. For many people, the only obvious symptom of sleep apnea is fatigue or excessive sleepiness during the day.
Other clues include waking up with a very dry mouth or sore throat (from breathing with your mouth open all night), frequent nighttime urination (sleep apnea can affect bladder sensation), difficulty concentrating, or mood changes. Some people even experience sexual dysfunction or decreased interest in sex due to the chronic sleep deprivation.
.The research suggests that some people may develop a heightened awareness over time, particularly if they also develop insomnia symptoms. Some patients with comorbid insomnia and sleep apnea may associate post-respiratory event awakenings with wakefulness, resulting in prolonged difficulty falling back asleep. In these cases, what started as unconscious micro-arousals can evolve into conscious awareness and anxiety about sleep disruption.
Why This All Matters
The health consequences of untreated sleep apnea extend far beyond feeling tired. Sleep apnea increases the risk of high blood pressure, irregular heart rhythms like atrial fibrillation, heart disease, and stroke. Each time your oxygen levels drop during a breathing pause, it stresses your cardiovascular system. Over months and years, this repeated stress can lead to serious complications.
There’s also the immediate danger of extreme daytime sleepiness. Excessive daytime sleepiness can cause microsleeps where you fall asleep for very brief periods—dangerous while driving or operating machinery. The cognitive fog, mood disturbances, and reduced productivity affect quality of life even if you never have a serious accident.
The good news is that treatment works. With proper diagnosis and appropriate therapy—whether that’s CPAP, an oral appliance, surgery, or lifestyle changes—most people see dramatic improvements in their sleep quality and daytime functioning. The key is recognizing the problem in the first place, which requires paying attention to those subtle clues your body provides during the day about what’s happening at night.
Medical Disclaimer
The information provided in this article is intended for general educational and informational purposes only and does not constitute medical advice. It should not be used as a substitute for professional medical advice, diagnosis, or treatment.
Always seek the guidance of a qualified healthcare provider with any questions you may have regarding a medical condition or treatment. Never disregard professional medical advice or delay seeking it because of something you have read here.
If you are experiencing a medical emergency, call 911 or your local emergency number immediately.
The author of this article is a licensed physician, but the views expressed here are solely those of the author and do not represent the official position of any hospital, health system, or medical organization with which the author may be affiliated.
Image generated by author using ChatGPT
Sources:
Mayo Clinic – Sleep Apnea: Symptoms and Causes
https://www.mayoclinic.org/diseases-conditions/sleep-apnea/symptoms-causes/syc-20377631
Cleveland Clinic – Sleep Apnea: What It Is, Causes, Symptoms & Treatment
https://my.clevelandclinic.org/health/diseases/8718-sleep-apnea
Merck Manual (Professional Edition) – Obstructive Sleep Apnea (OSA)
https://www.merckmanuals.com/professional/pulmonary-disorders/sleep-apnea/obstructive-sleep-apnea-osa
StatPearls/NCBI – Obstructive Sleep Apnea
https://www.ncbi.nlm.nih.gov/books/NBK459252/
American Lung Association – Obstructive Sleep Apnea Symptoms and Diagnosis
https://www.lung.org/lung-health-diseases/lung-disease-lookup/sleep-apnea/symptoms-diagnosis







Trump, MAGA, and the Politics of Permission
By John Turley
On July 18, 2026
In Commentary, Politics
Donald Trump did not invent a new American bigotry. He did something more subtle—and perhaps more consequential. He transformed long-standing resentments into a political identity and gave many people permission to express views they had previously kept private.
The central question surrounding the Make America Great Again movement is: has it created a new ethical worldview, or has it simply exposed attitudes that have long existed beneath the surface of American society? The answer is probably: both.
The resentments themselves are not new. Long before Trump entered politics, historians and political scientists documented the enduring influence of racial resentment, nativism, religious nationalism, and cultural anxiety. Many Americans believed their country was changing too quickly and that immigration, globalization, and shifting social norms threatened a way of life they once took for granted.
American history offers countless examples. Anti-immigrant movements flourished in the nineteenth century. Segregation and white supremacy were embedded in law for generations. George Wallace’s “law and order” campaigns, Pat Buchanan’s culture-war speeches, and the Tea Party’s reaction to Barack Obama’s presidency all reflected recurring fears that “real America” was slipping away.
Obama’s election encouraged hopes that the nation had entered a post-racial era, yet research continued to show that racial and cultural anxieties remained powerful undercurrents. Trump did not create those sentiments. He recognized them, embraced them, and gave them a simple narrative.
“Make America Great Again” is more than a campaign slogan. It tells a story of loss, restoration, and retribution. It suggests that something valuable has been taken away and that reclaiming it is both necessary and morally justified. For many supporters, it became less about policy than identity.
Trump’s most significant innovation was not ideological but cultural. Previous politicians often relied on coded language while preserving at least the appearance of restraint. Trump discarded those conventions. He mocked opponents, encouraged inflammatory chants, proposed banning Muslims from entering the country, and described immigrants as “poisoning the blood” of America. Many supporters interpreted this not as recklessness but as authenticity—a rejection of what they viewed as the constraints of political correctness.
That shift changed more than political rhetoric. It altered the boundaries of acceptable public behavior. Conduct once viewed as disqualifying became, for many supporters, evidence of courage and honesty. Loyalty increasingly outweighed character. Offending critics became proof that one was willing to stand up to elite opinion.
Political scientists describe this process as norm erosion. Laws remain the same, but the unwritten rules governing public life gradually weaken. Loyalty becomes more important than truth, institutions are trusted only when they favor one’s own side, and democratic restraints come to be seen as obstacles rather than safeguards.
Trump did not invent this dynamic. Populist movements throughout history have divided society into virtuous insiders and corrupt outsiders. His contribution was to make that style of politics a daily spectacle. Political conflict became a struggle between patriots and traitors, real Americans and dangerous enemies. Once politics is framed in existential terms, compromise becomes betrayal and restraint begins to look like weakness.
It would be inaccurate to say Trump created prejudice from nothing. A more persuasive argument is that he reduced the social cost of expressing it. Social norms matter because they define the boundaries of acceptable conduct. When prominent public figures use inflammatory language or repeat conspiracy theories without losing support from their followers, those boundaries inevitably shift.
Supporters who might once have hesitated can now dismiss criticism as media bias or political correctness. The line between prejudice and plain speaking becomes increasingly blurred.
This may prove to be Trump’s most enduring legacy. Even if his personal political influence fades, the standards governing public discourse may not return to where they once were. Future politicians can imitate the style—its confrontation, norm-breaking, and appeals to grievance—even if they reject parts of Trump’s agenda.
That is why the distinction between creating prejudice and legitimizing it matters. If Trump merely invented these impulses, defeating one political leader might solve the problem. But if he tapped into deeper currents already present in American society, then the challenge extends far beyond one election.
Trump is best understood as both symptom and accelerant. He gave shape to existing resentments, transformed them into a powerful political identity, and lowered the barriers against openly expressing them. The prejudices themselves were not new. The confidence with which they could be publicly displayed—and sometimes celebrated—was.
Whether that change proves temporary or enduring may become one of the defining questions facing American democracy.